生物大分子的功能主要由其构象转变所驱动。阐明生物大分子在不同构象状态下的结构,能有效帮助我们理解这些分子发挥功能的结构基础。在各种技术中,冷冻电镜(cryo-EM)已被证明是一种强大的工具,能够以原子或近原子分辨率对生物大分子进行结构解析。这项技术能够在毫秒内快速玻璃化生物样本,故而理论上能捕捉生物大分子在不同构象状态下的结构。
然而,根据动态平衡定律(law of dynamic equilibrium),生物大分子处于不同构象状态的占比存在显著差异。在许多案例中,处于低能态的常见构象在生物大分子颗粒中的数量较多,因而能够获得高分辨率的密度图;而稀有构象处于较高能量的准稳定状态,所以在平衡条件下颗粒稀少。尽管后者对揭示关键生物过程至关重要,却往往难以达到满意的分辨率。如何提升这些稀有构象的分辨率,是冷冻电镜领域长期面临的挑战。

2024年8月28日,胡名旭特聘研究员、包承龙副教授以及史作强教授在《通讯·生物》(Communications Biology)杂志上共同发表标题为 CryoTRANS: Predicting High-resolution Maps of Rare Conformations from Self-supervised Trajectories in Cryo-EM 的研究论文,提出一种基于自监督轨迹的预测冷冻电镜稀有构象高分辨率密度图的算法。
CryoTRANS是一种基于自监督方式训练神经网络的优化模型,它通过深度神经网络参数化的常微分方程,模拟生物大分子从高分辨率常见构象到低分辨率稀有构象的连续形变过程。CryoTRANS使用了 Wasserstein 距离(即最优传输距离)来度量“预测密度图”和“目标低分辨率密度图”的差异,并对速度场施加了刚性约束,使得初始常见构象的高分辨率细节在整个形变过程中得以保持。因此,形变生成的密度图可以视为目标低分辨率结构的高分辨率预测。

图1. CryoTRANS的模型结构和基本原理
CryoTRANS 生成密度图的质量和准确性在四套模拟数据集上得到了验证。这些数据集各自包含两种不同的构象状态,其中一个构象的原子模型被低通滤波至3Å,作为CryoTRANS的初始密度图,另一个构象的原子模型被分别低通滤波至3Å,5Å 和7Å作为目标密度图。结果显示,当初始密度图和目标密度图的分辨率均为 3Å 时,CryoTRANS生成的密度分辨率高于3.2Å(注:本文所提及的分辨率均为模型-密度图分辨率(model-to-map resolution),即model-to-map FSC取0.5为阈值下的频率);当目标图的分辨率为5Å 或 7Å 时,生成的密度能保留约4Å 的信息。这表明,经由深度神经网络学习得到的速度场,CryoTRANS能够通过保留初始密度的高分辨率结构细节。
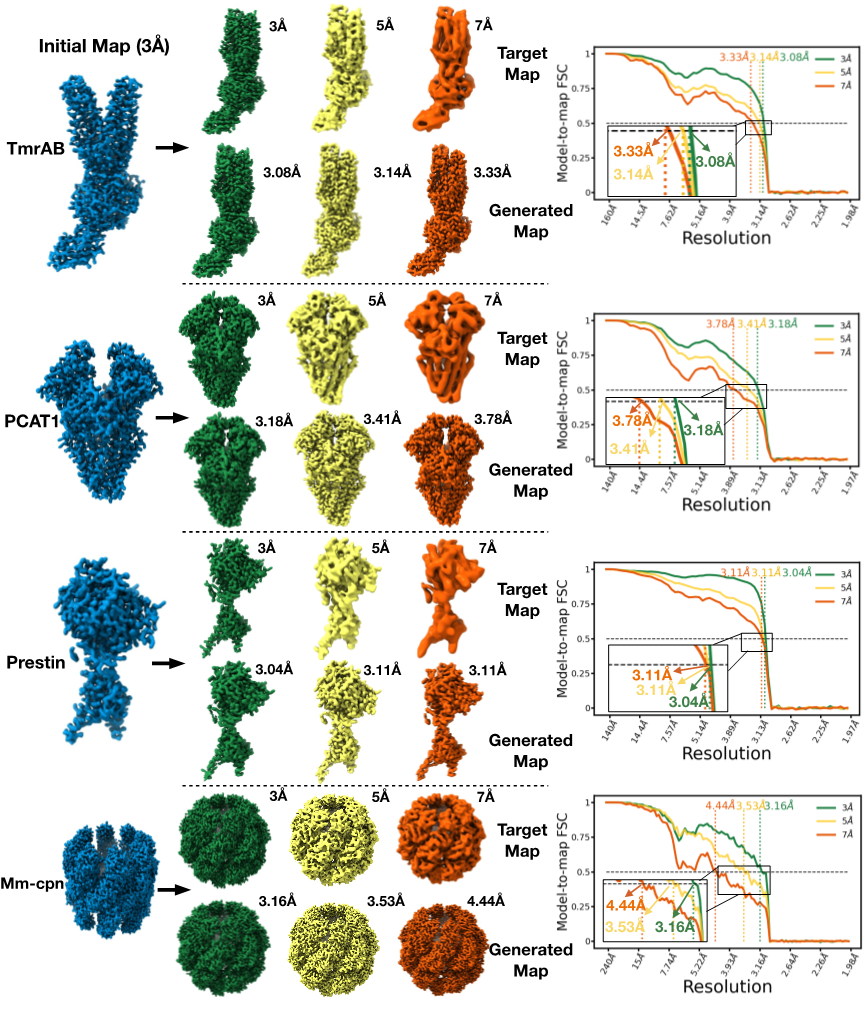
图2. CryoTRANS预测结果的准确性(模拟数据集)
除了模拟数据集外,CryoTRANS 也在三个冷冻电镜公开数据集上验证了其有效性:α2-巨球蛋白(A2M)、肌动蛋白结合蛋白复合物(Arp2/3)以及70S核糖体。
实验结果表明,CryoTRANS能够生成准确预测稀有构象高分辨率结构的密度图。例如,A2M在induced state和native state下分辨率分别为4.39Å和8.02Å,native state的低分辨率对原子模型构建构成挑战。CryoTRANS生成了A2M从induced state转变为native state的高质量形变轨迹,将native state的分辨率提高到了4.84 Å。对于Arp2/3,因其柔性导致目标构象中存在部分缺失的密度,CryoTRANS 生成的密度图能够有效补充这些缺失的部分,并揭示了许多原本缺失的高分辨率细节,分辨率可达 3.79 Å,远高于目标图的 9.82 Å。
此外,与基于深度学习的后处理方法(如DeepEMhancer和EMReady)相比,CryoTRANS 生成图的分辨率显著更优。CryoTRANS的另一大优点在于,它使用的是自监督方法,无需在大型数据集上进行预训练,因此不受模型偏差的影响。

图3. CryoTRANS预测了A2M和Arp2/3稀有构象的高分辨率结构
CryoTRANS 展现了架接冷冻电镜单颗粒分析(SPA)与冷冻电子断层扫描(cryo-ET)这两种技术的潜力。利用高分辨率的 SPA 密度作为起点,CryoTRANS 能够有效地从低分辨率的 cryo-ET 密度中预测出高分辨率结构。以 SARS-CoV-2 刺突蛋白为例,初始构象来自 cryo-EM SPA (EMDB-22001),其分辨率为 3.57 Å,对应三个受体结合域(RBD)处于闭合状态;目标构象则来自cryo-ET(EMDB-16697),分辨率为 13.2 Å,对应一个 RBD 处于开放状态。从3.57 Å 的SPA构象出发,CryoTRANS 成功模拟了 RBD 区域的旋转,同时生成了3.78 Å的目标构象,这验证了 CryoTRANS 利用 SPA 获得的结构来提升 cryo-ET 密度分辨率的能力,为不同成像模式之间的跨模态融合提供了重要的洞察。
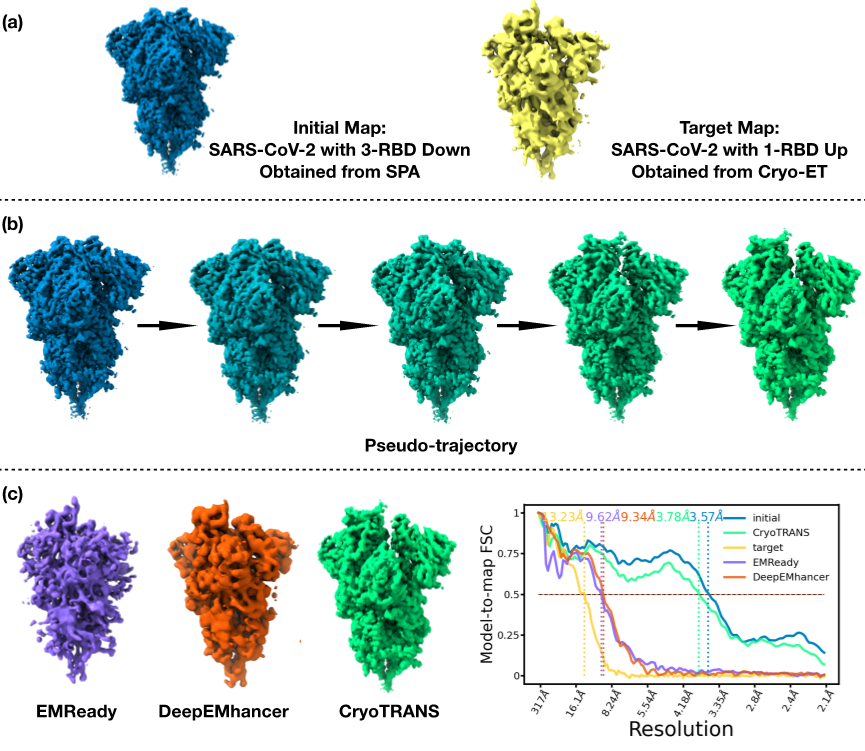
图4. CryoTRANS使用SPA密度预测cryo-ET密度的高分辨率结构
目前,CryoTRANS已经在Github上发布,可点击链接 https://github.com/mxhulab/cryotrans下载软件。
胡名旭课题组 现面向社会公开招聘副研究员、高级工程师、博士后和助理研究员,欢迎加入!
清华大学丘成桐数学中心副教授、北京雁栖湖应用数学研究院助理研究员、清华大学膜生物学重点实验室研究员包承龙,深圳医学科院特聘研究员、北京市生物结构前沿中心(清华大学)研究员胡名旭,清华大学丘成桐数学中心教授、北京雁栖湖应用数学研究院研究员史作强为本文的共同通讯作者。清华大学丘成桐数学中心博士研究生樊箫,清华大学生命科学院博士研究生张起为本文的共同第一作者。清华大学求真书院博士生张慧,清华大学丘成桐数学中心博士生祝健颖,及美国南卡莱罗纳大学教授鞠立力为共同作者。
