冷冻电子显微镜(Cryogenic electron microscopy, cryo-EM)单颗粒分析(single-particle analysis, SPA)是确定生物大分子近原子分辨率结构时广泛使用的技术方法。通过透射电子显微镜,研究者可以记录单个大分子颗粒在不同投影角度下的二维投影图像,进而重构三维电势密度图,搭建分子结构模型。然而,当生物大分子颗粒数较少(或者是蛋白表达量低,或者是高能中间态极其罕见)或出现角度缺失(如优势取向)的情况时,重构的密度图质量变差,导致结构解析精度受限。
北京时间2026年1月19日,包承龙副教授、胡名旭特聘研究员在《通讯·化学》(Communications Chemistry)杂志上共同发表了题为“Fine-tuning AlphaFold with limited cryo-EM observations”的研究论文。该研究提出了一种名为CoCoFold的端到端微调框架,通过将冷冻电镜原始颗粒图像直接整合进AlphaFold的结构预测流程中,实现了在极少量观测数据下的高精度原子模型预测。

尽管AlphaFold在蛋白质结构预测上取得了巨大成功,但其预测结果仍可能与实验观测存在偏差,特别是在处理具有多种构象或同源信息不足的蛋白质时。传统的冷冻电镜模型构建方法(如Phenix、ModelAngelo)高度依赖于高质量的密度图。然而,当面临以下两种“极限挑战”时,这些方法的表现往往会大幅下降:
1. 颗粒稀少:例如内源性蛋白表达量低,或处于低概率高能态的蛋白构象,难以收集到海量图像。
2. 视角缺失:由于蛋白质在气液界面的吸附,导致颗粒倾向于某些特定角度,从而造成重构密度图的严重各向异性。
CoCoFold的核心思路是:不再依赖重构后的密度图,而是直接利用原始颗粒图像来微调AlphaFold预训练权重。
其架构设计具有以下亮点:
内存高效的微调策略:研究团队冻结了AlphaFold 的Evoformer模块,仅微调其结构模块(Structure Module)。通过引入融合注意力机制(fused attention),CoCoFold能够在不显著增加计算负担的情况下,将图像信息引导至模型预测过程中。
端到端可微链接:CoCoFold包含一个可微的“高斯混合MolMap”模块。该模块将预测的原子坐标转化为模拟密度图,并生成2D投影图,直接与实验观察到的原始颗粒图像进行对比(基于傅里叶环相关系数损失函数),从而实现端到端的参数更新。
物理先验的保持:通过从预训练的AlphaFold权重开始微调,CoCoFold既能吸收实验数据提供的局部约束,又能利用AlphaFold学习到的蛋白质结构物理先验,防止模型在极少数据下产生非物理的形变。
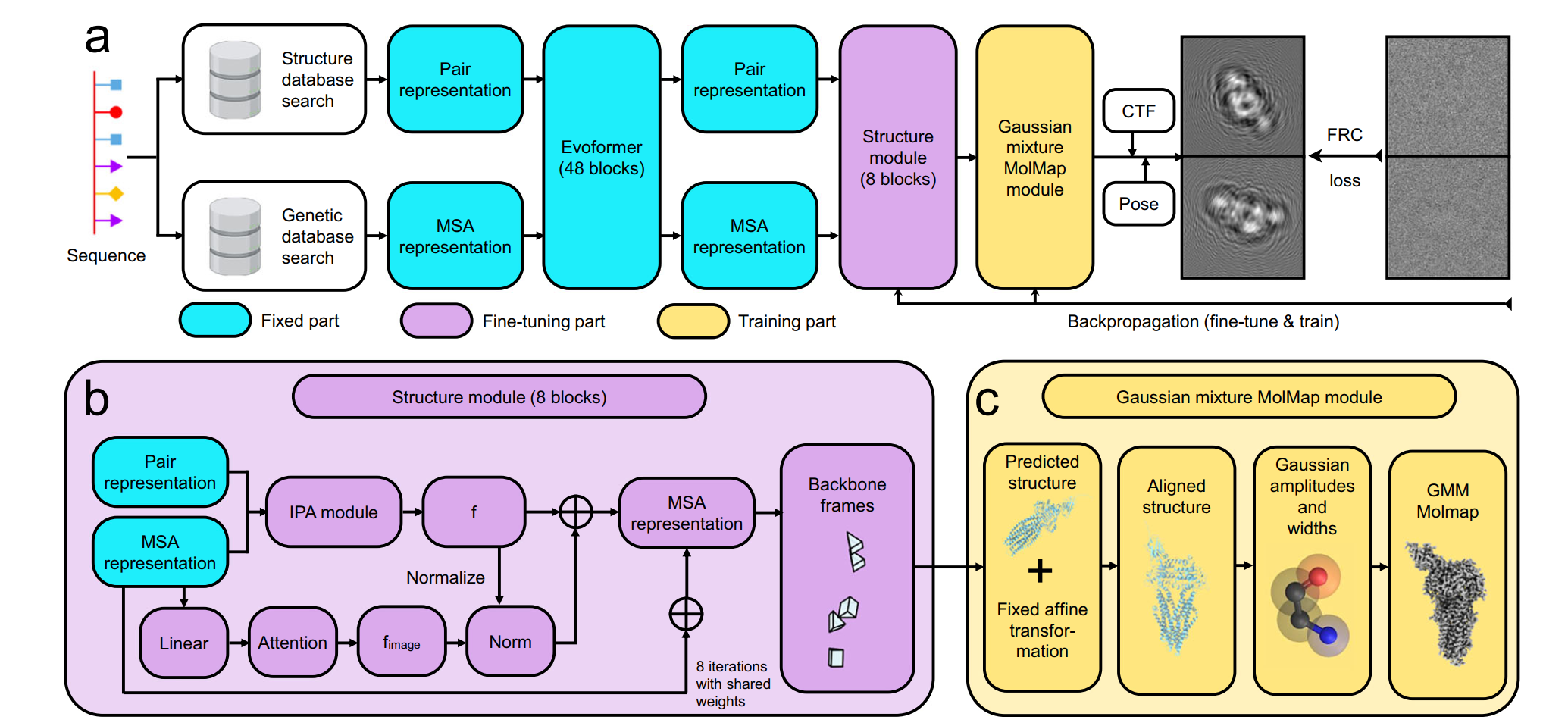
图注:CoCoFold算法框架
在传统的冷冻电镜工作流中,研究者通常遵循“2D颗粒提取 -> 3D密度图重构 -> 原子模型构建”的线性步骤。然而,这种流程在面对“极端数据”时存在一个“重构陷阱”:当颗粒数量极少或视角严重缺失时,3D重构算法会产生严重的伪影(如拉长或者模糊)。如果模型构建工具(如ModelAngelo[1])仅依赖于这些“失真”的密度图,预测结果会偏离真实结构。CoCoFold 的创新之处在于它“绕过了中间商”。它直接利用2D颗粒图像作为约束,通过可微的投影算子,在AlphaFold的预测空间与原始实验观测空间之间架起了一座桥梁。这意味着,即使3D密度图已经模糊到肉眼无法辨识,CoCoFold依然能从2D信号中捕捉到细微的结构特征,从而纠正AlphaFold初始预测中的偏差。
研究团队在多个实验和模拟数据集上进行了压力测试,并与DiffModeler、ModelAngelo、MICA等5种前沿方法进行了对比,与传统方法对比取得明显优势。在最极端的情况下,利用1.1K 颗粒便可将AlphaFold预测的RMSD大于5 Å(与真实结构对比)的结构微调至2 Å。
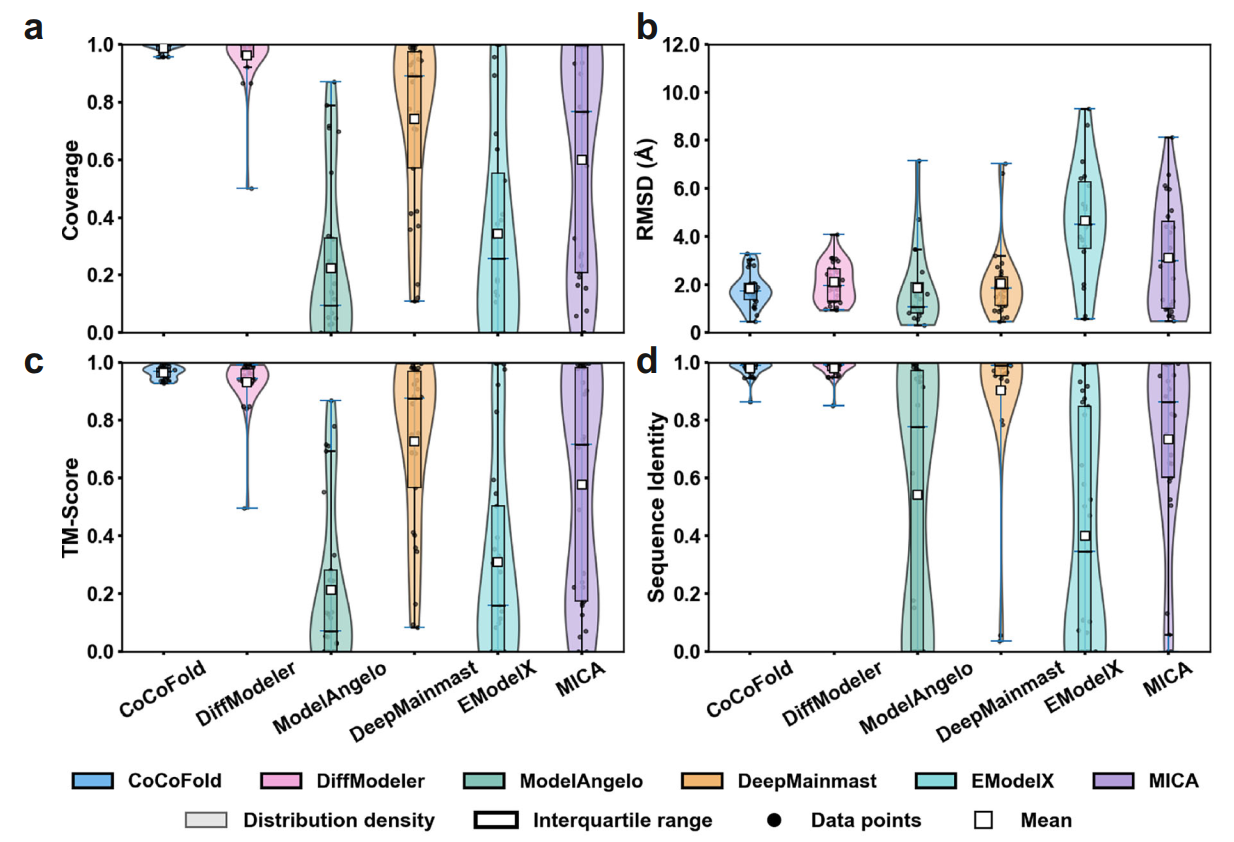
图注:有限颗粒数下不同方法对比
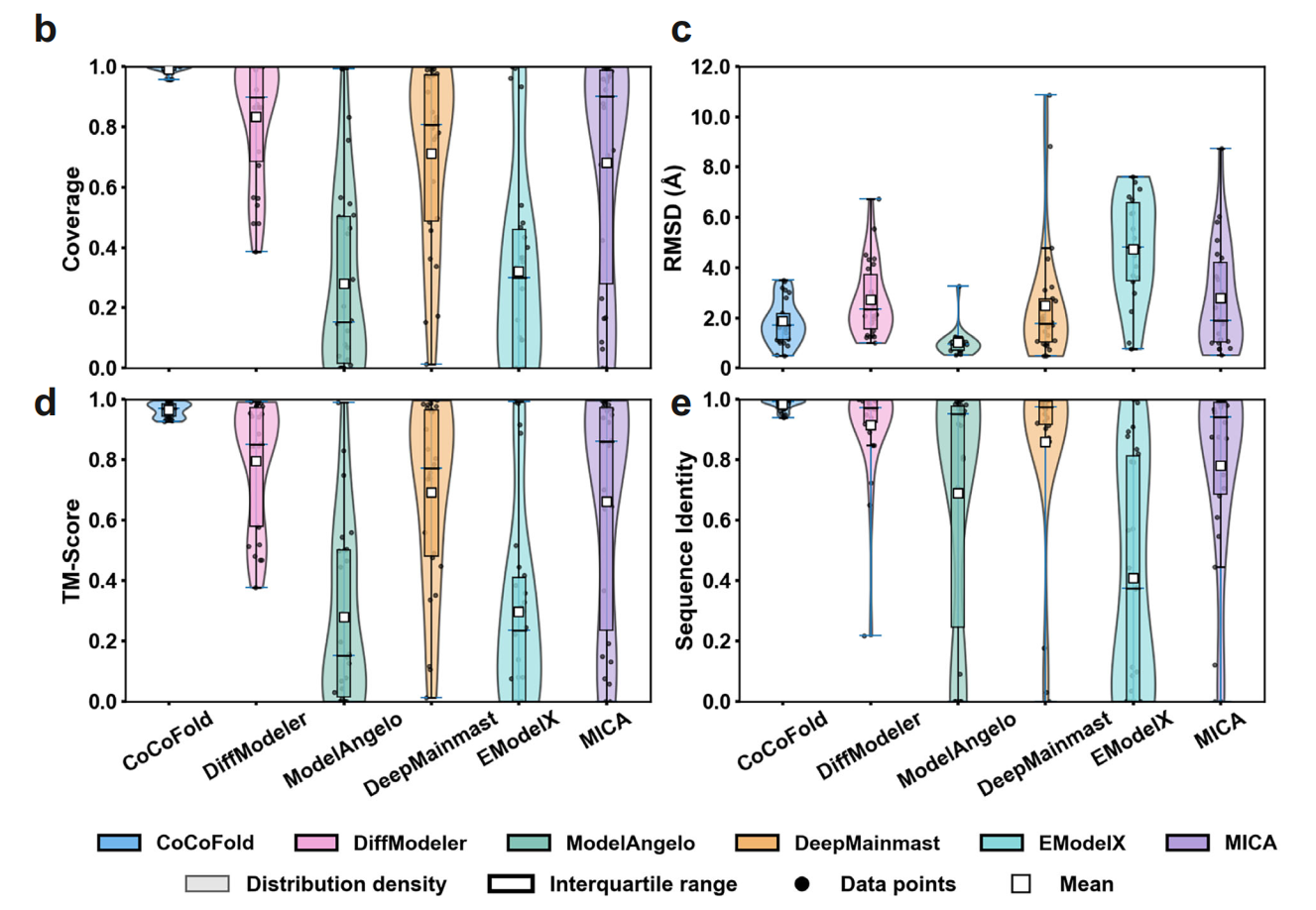
图注:有限观测角度下不同方法对比
此外,研究者进一步在MSP-1蛋白质上对比了使用1.1K 原始颗粒与其重建密度图的重投影颗粒对AlphaFold的微调效果。结果表明前者效果显著好于后者。重构重建密度图本质上是一个平均过程,会损失高频信息。CoCoFold直接从原始颗粒中学习,保留了更多细节。
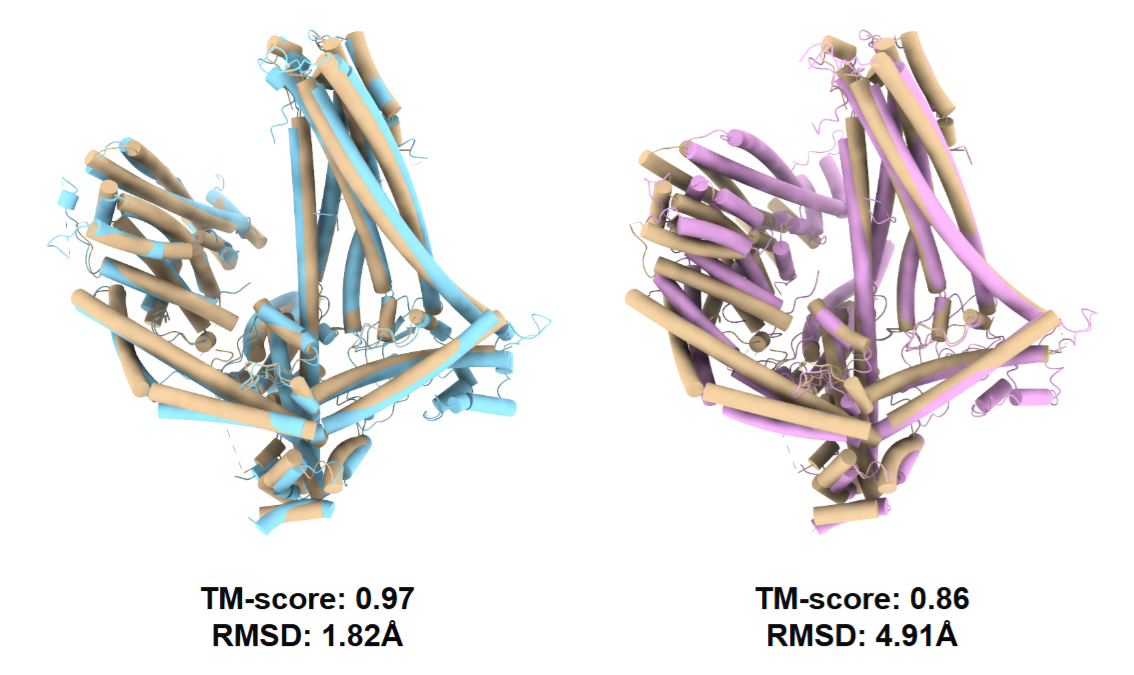
图注:黄色为真实结构,左侧蓝色为基于真实原始颗粒微调结构,右侧粉色为基于密度图重投影颗粒微调结构
对于含有大量数据但想节省计算成本的用户,研究者的实验还表明:利用CryoSieve[2]筛选出少量(如3000个)高质量粒子,再跑CoCoFold,仅需20多分钟就能获得精准的结构 。
清华大学丘成桐数学中心副教授、北京雁栖湖应用数学研究院研究员、清华大学膜生物学重点实验室研究员包承龙和深圳医学科学院特聘研究员胡名旭为本文的共同通讯作者。清华大学求真书院博士研究生廖俊文、张慧和清华大学丘成桐数学中心博士研究生郑棣瀚(已毕业)为本文的共同第一作者。本研究受到深圳医学科学院特聘研究员启动经费、北京市生物结构前沿中心(清华大学)、国家自然科学基金、国家重点研发计划的资助。
参考文献:
[1] Jamali, K., Käll, L., Zhang, R., Brown, A., Kimanius, D., & Scheres, S. H. (2024). Automated model building and protein identification in cryo-EM maps. Nature, 628(8007), 450-457.
[2] Liao, J., Zheng, D., Zhang, H. et al. (2026). Fine-tuning AlphaFold with limited cryo-EM observations. Commun Chem 9, 95.
talent@smart.org.cn
researcher@smart.org.cn
recruitment@smart.org.cn
graduate_office@smart.org.cn
graduate_admission@smart.org.cn
otl@smart.org.cn
smartfund@smart.org.cn
pr@smart.org.cn
