Cryogenic electron microscopy (cryo-EM) single-particle analysis (SPA) is a widely used technique for determining near-atomic-resolution structures of biological macromolecules. Through transmission electron microscopy, researchers can record two-dimensional projection images of individual macromolecular particles at different projection angles, thereby reconstructing a three-dimensional electric potential density map to build molecular structural models. However, when the number of biological macromolecular particles is small (either due to low protein expression or extremely rare high-energy intermediate states) or when there is missing angle information (such as preferred orientation), the quality of the reconstructed density map deteriorates, limiting the precision of structural resolution.
On January 19, 2026 (Beijing Time), Associate Professor Chenglong Bao and Junior PI Mingxu Hu jointly published a research paper titled "Fine-tuning AlphaFold with limited cryo-EM observations" in the journal Communications Chemistry. This study proposes an end-to-end fine-tuning framework named CoCoFold. By directly integrating raw cryo-EM particle images into AlphaFold’s structure prediction pipeline, it achieves high-precision atomic model prediction with extremely limited observation data.

Although AlphaFold has achieved tremendous success in protein structure prediction, its predictions may still deviate from experimental observations, especially for proteins with multiple conformations or lacking homologous information. Traditional cryo-EM model-building methods (such as Phenix and ModelAngelo) highly depend on high-quality density maps. However, when facing the following two "extreme challenges", the performance of these methods often drops significantly:
Scarcity of Particles: For instance, low expression of endogenous proteins or protein conformations in low-probability, high-energy states makes it difficult to collect a massive number of images.
Missing Views: Due to the adsorption of proteins at the air-water interface, particles tend to adopt certain specific angles, resulting in severe anisotropy in the reconstructed density maps.
The core idea of CoCoFold is to bypass the reliance on reconstructed density maps and instead directly utilize raw particle images to fine-tune the pre-trained weights of AlphaFold.
Its architectural design features the following highlights:
Memory-efficient fine-tuning strategy: The research team froze the Evoformer module of AlphaFold and only fine-tuned its Structure Module. By introducing a lightweight attention adapter (fused attention), CoCoFold can guide image information into the model prediction process without significantly increasing the computational burden.
End-to-end differentiable link: CoCoFold includes a differentiable "Gaussian Mixture MolMap" module. This module converts predicted atomic coordinates into simulated density maps and generates 2D projections, which are directly compared with the experimentally observed raw particle images (based on a Fourier Ring Correlation loss function), thereby enabling end-to-end parameter updates.
Preservation of physical priors: By starting the fine-tuning from pre-trained AlphaFold weights, CoCoFold can absorb the local constraints provided by experimental data while leveraging the physical priors of protein structures learned by AlphaFold. This prevents the model from generating non-physical deformations under extremely sparse data conditions.
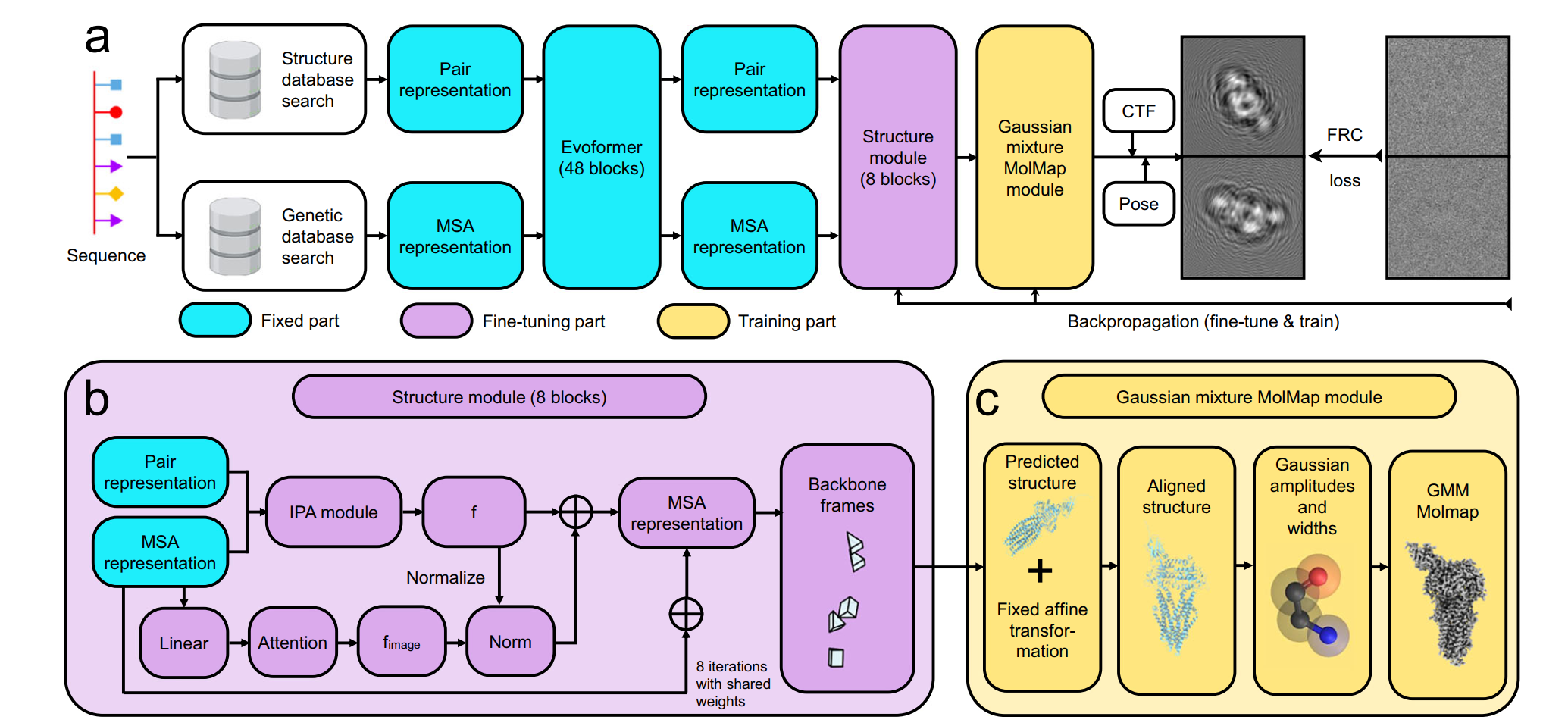
The CoCoFold algorithm framework
In traditional cryo-EM workflows, researchers typically follow the linear steps of "2D particle extraction -> 3D density map reconstruction -> atomic model building". However, this pipeline faces a "reconstruction trap" when dealing with "extreme data": when the number of particles is extremely low or views are severely missing, 3D reconstruction algorithms generate severe artifacts (such as elongation or blurring). If model-building tools (such as ModelAngelo[1]) solely rely on these "distorted" density maps, the predicted results will deviate from the true structure.
The innovation of CoCoFold lies in its ability to "bypass the middleman". It directly uses 2D particle images as constraints, building a bridge between AlphaFold’s prediction space and the raw experimental observation space via a differentiable projection operator. This means that even if the 3D density map is too blurry to be recognizable by the naked eye, CoCoFold can still capture subtle structural features from the 2D signals, thereby correcting biases in AlphaFold’s initial predictions.
The research team conducted stress tests on multiple experimental and simulated datasets and compared CoCoFold with five cutting-edge methods, including DiffModeler, ModelAngelo, and MICA, achieving significant advantages over traditional methods. In the most extreme case, using only 1.1K particles, it was able to fine-tune a structure predicted by AlphaFold with an RMSD greater than 5 Å (compared to the true structure) down to 2 Å.
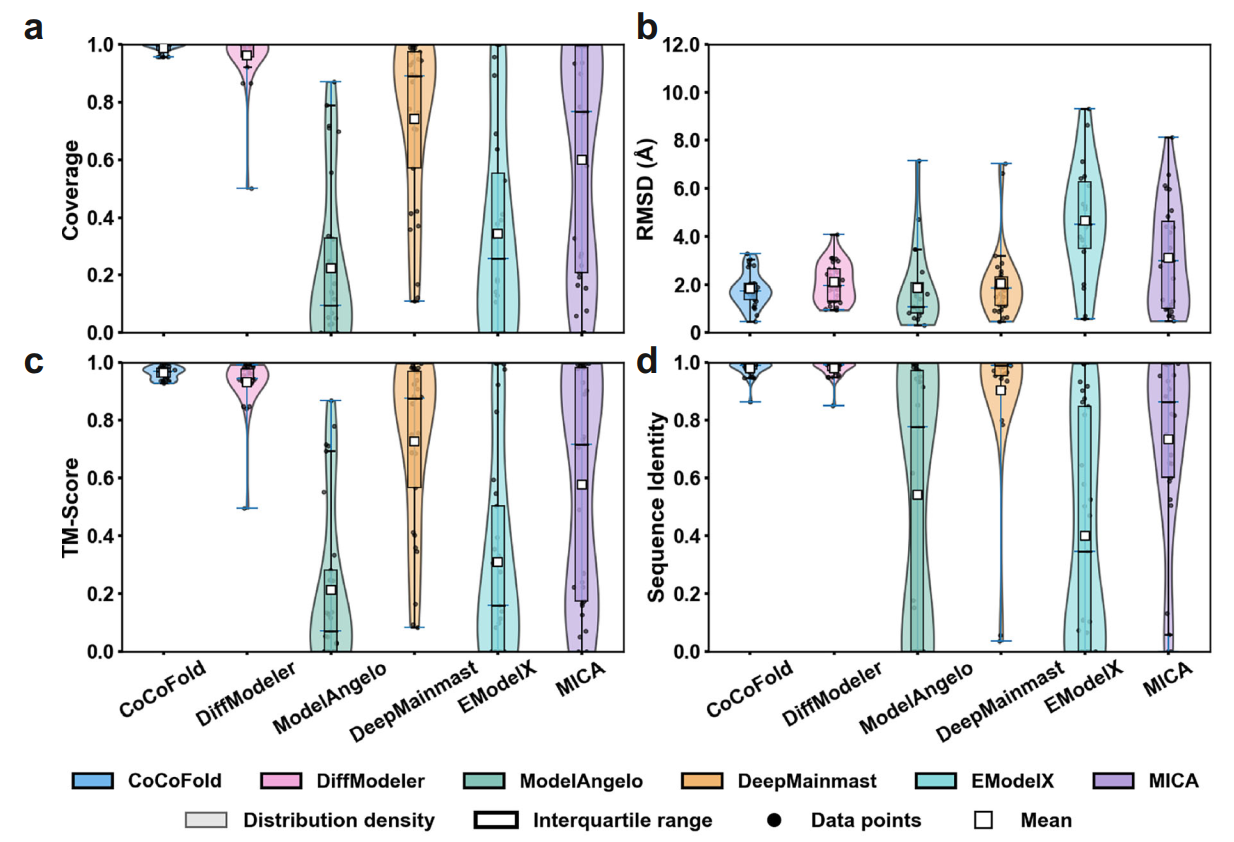
Comparison of different methods under limited particle counts
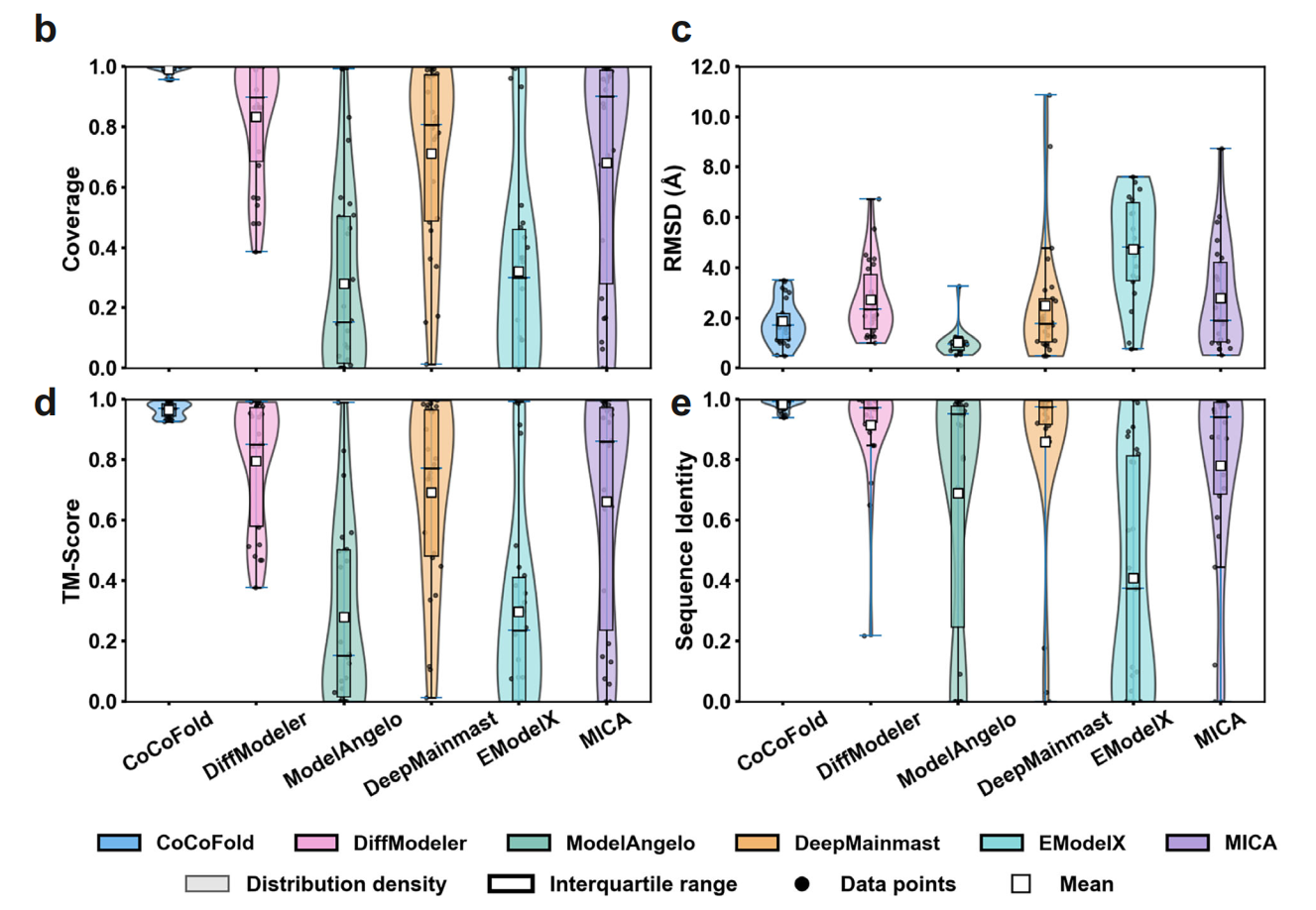
Comparison of different methods under limited observation angles
Furthermore, researchers evaluated the fine-tuning effects on AlphaFold using 1.1K raw particles versus re-projected particles from their reconstructed density maps on the MSP-1 protein. The results showed that the former performed significantly better than the latter. Reconstructing a density map is essentially an averaging process, which leads to the loss of high-frequency information. CoCoFold learns directly from the raw particles, preserving more details.
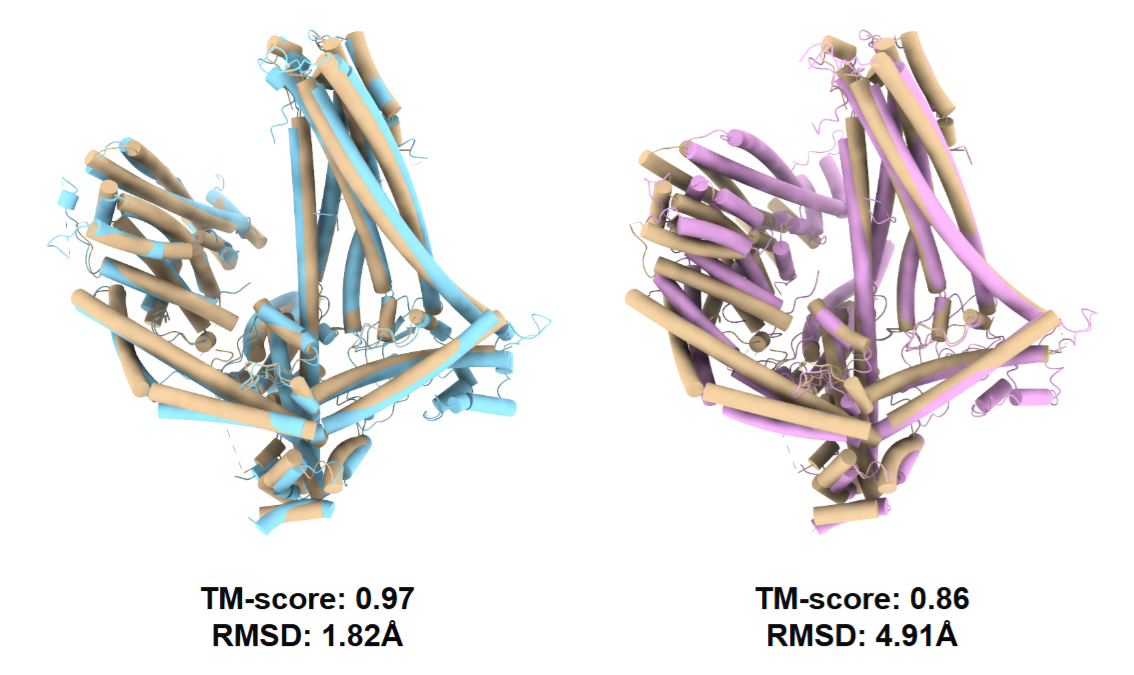
Yellow indicates the true structure; blue on the left indicates the fine-tuned structure based on real raw particles; pink on the right indicates the fine-tuned structure based on density map re-projected particles
For users who have massive amounts of data but wish to save computational costs, the researchers’ experiments also demonstrated that by using CryoSieve[2] to filter out a small number of high-quality particles (e.g., 3,000), running CoCoFold only takes over 20 minutes to obtain an accurate structure.
Associate Professor Chenglong Bao from the Yau Mathematical Sciences Center at Tsinghua University, PI at the Yanqi Lake Beijing Institute of Mathematical Sciences and Applications, and PI at the State Key Laboratory of Membrane Biology at Tsinghua University, along with Junior PI Mingxu Hu from the Shenzhen Medical Academy of Research and Translation (SMART), are the co-corresponding authors of this paper. PhD. Student Junwen Liao and Hui Zhang from Qiuzhen College at Tsinghua University, and PhD. Student Dihan Zheng (graduated) from the Yau Mathematical Sciences Center at Tsinghua University is the co-first author. This research was funded by the Junior PI Start-up Fund of the Shenzhen Medical Academy of Research and Translation, the Beijing Advanced Innovation Center for Structural Biology (Tsinghua University), the National Natural Science Foundation of China, and the National Key Research and Development Program.
References:
[1] Jamali, K., Käll, L., Zhang, R., Brown, A., Kimanius, D., & Scheres, S. H. (2024). Automated model building and protein identification in cryo-EM maps. Nature, 628(8007), 450-457.
[2] Liao, J., Zheng, D., Zhang, H. et al. (2026). Fine-tuning AlphaFold with limited cryo-EM observations. Commun Chem 9, 95.
Translation: Yang Shen
talent@smart.org.cn
researcher@smart.org.cn
recruitment@smart.org.cn
graduate_office@smart.org.cn
graduate_admission@smart.org.cn
otl@smart.org.cn
smartfund@smart.org.cn
pr@smart.org.cn
Subscription successful! Thank you for following SMART.
